Рецепты мяса по-французски в духовке с мясом, грибами, картошкой
Мясо по-французски - как много в этом слове для сердца русского слилось… Несмотря на название, это блюдо уже давно стало...
СПЕКТРАЛЬНЫЙ АНАЛИЗ
,
метод качеств. и количеств. определения состава в-в, основанный на исследовании
их спектров испускания, поглощения, отражения и люминесценции . Различают атомный
и молекулярный спектральный анализ, задачи к-рых состоят в определении соотв. элементного
и молекулярного состава в-ва. Эмиссионный спектральный анализ проводят по спектрам испускания
атомов , ионов или молекул , возбужденных разл. способами, абсорбционный спектральный анализ-по
спектрам поглощения электромагн. излучения анализируемыми объектами (см. Абсорбционная
спектроскопия). В зависимости от цели исследования, св-в анализируемого
в-ва, специфики используемых спектров, области длин волн и др. факторов ход
анализа, аппаратура, способы измерения спектров и метро-логич. характеристики
результатов сильно различаются. В соответствии с этим спектральный анализ подразделяют на
ряд самостоят. методов (см., в частности, Атомно-абсорбционный анализ , Атомно-флуоресцентный
анализ , Инфракрасная спектроскопия , Комбинационного рассеяния спектроскопия ,
Люминесцентный анализ , Молекулярная оптическая спектроскопия , Спектроскопия
отражения , Спектрофотометрия , Ультрафиолетовая спектроскопия , Фотометрический
анализ , Фурье-спектроскопия , Рентгеновская спектроскопия).
Часто под спектральным анализом понимают
только атомно-эмис-сионный спектральный анализ (АЭСА)-метод элементного анализа ,
основанный на изучении спектров испускания
своб. атомов и ионов в газовой фазе в области длин волн 150-800 нм (см. Атомные
спектры).
При анализе твердых в-в
наиб. часто применяют дуговые (постоянного и переменного тока) и искровые разряды,
питаемые от специально сконструир. стабилизир. генераторов (часто с электронным
управлением). Созданы также универсальные генераторы, с помощью к-рых получают
разряды разных типов с переменными параметрами, влияющими на эффективность процессов
возбуждения исследуемых образцов. Твердая электропроводящая проба непосредственно
может служить электродом дуги или искры; не проводящие ток твердые пробы и порошки
помещают в углубления угольных электродов той или иной конфигурации. В этом
случае осуществляют как полное испарение (распыление) анализируемого в-ва, так
и фракционное испарение последнего и возбуждение компонентов пробы в соответствии
с их физ. и хим. св-вами, что позволяет повысить чувствительность и точность
анализа. Для усиления эффекта фракционирования испарения широко применяют добавки
к анализируемому в-ву реагентов , способствующих образованию в условиях высокотемпературной
[(5-7)·10 3 К] угольной дуги легколетучих соед. (фторидов , хлоридов ,
сульфидов и др.) определяемых элементов. Для анализа геол. проб в виде порошков
широко применяют способ просыпки или вдувания проб в зону разряда угольной дуги.
При анализе металлургия ,
проб наряду с искровыми разрядами разных типов используют также источники света
тлеющего разряда (лампы Грима, разряд в полом катоде). Разработаны комбинир.
автоматизир. источники, в к-рых для испарения или распыления используют лампы
тлеющего разряда или электротермич. анализаторы, а для получения спектров, напр.,-высокочастотные
плазматроны. При этом удается оптимизировать условия испарения и возбуждения
определяемых элементов.
При анализе жидких проб
(р-ров) наилучшие результаты получаются при использовании высокочастотных (ВЧ)
и сверхвысокочастотных (СВЧ) плазматронов, работающих в инертной атмосфере ,
а также при пламенно-фотометрич. анализе (см. Фотометрия пламени эмиссионная).
Для стабилизации т-ры плазмы разряда на оптимальном уровне вводят добавки
легкоионизируемых в-в, напр. щелочных металлов . Особенно успешно применяют ВЧ
разряд с индуктивной связью тороидальной конфигурации (рис. 1). В нем разделены
зоны поглощения ВЧ энергии и возбуждения спектров, что позволяет резко повысить
эффективность возбуждения и отношение полезного аналит. сигнала к шуму и, т.
обр., достичь очень низких пределов обнаружения широкого круга элементов. В
зону возбуждения пробы вводят с помощью пневматических или (реже) ультразвуковых
распылителей. При анализе с применением ВЧ и СВЧ плазматронов и фотометрии пламени
относит. стандартное отклонение составляет 0,01-0,03, что в ряде случаев позволяет
применять АЭСА вместо точных, но более трудоемких и длительных хим. методов
анализа.
Для анализа газовых смесей
необходимы спец. вакуумные установки; спект-ры возбуждают с помощью ВЧ и СВЧ
разрядов. В связи с развитием газовой хроматографии эти методы применяют редко.
Рис. 1. ВЧ плазматрон:
1-факел отходящих газов ; 2-зона возбуждения спектров; 3-зона поглощения ВЧ энергии;
4-нагреват. индуктор; 5-вход охлаж-дающега газа (азот , аргон); 6-вход плазмообра-зующего
газа (аргон); 7-вход распыленной пробы (несущий газ-аргон).
При анализе в-в высокой
чистоты, когда требуется определять элементы, содержание к-рых меньше 10 -5 -10
%, а также при анализе токсичных и радиоактивных в-в пробы предварительно обрабатывают;
напр., частично или полностью отделяют определяемые элементы от основы и переводят
их в меньший объем р-ра или вносят в меньшую массу более удобного для анализа
в-ва. Для разделения компонентов пробы применяют фракционную отгонку основы
(реже-примесей), адсорбцию , осаждение , экстракцию , хроматографию , ионный обмен .
АЭСА с использованием перечисленных хим. способов концентрирования пробы , как
правило, наз. химико-спектральным анализом. Дополнит. операции разделения и
концентрирования определяемых элементов заметно повышают трудоемкость и длительность
анализа и ухудшают его точность (относит. стандартное отклонение достигает значений
0,2-0,3), но снижает пределы обнаружения в 10-100 раз.
Специфич. областью АЭСА
является микроспектральный (локальный) анализ. При этом микрообъем в-ва (глубина
кратера от десятков мкм до неск. мкм) испаряют обычно лазерным импульсом, действующим
на участок пов-сти образца диаметром неск. десятков мкм. Для возбуждения спектров
используют чаще всего импульсный искровой разряд, синхронизованный с лазерным
импульсом. Метод применяют при исследовании минералов , в металловедении.
Спектры регистрируют с
помощью спектрографов и спектрометров (квантометров). Имеется много типов этих
приборов, различающихся светосилой, дисперсией, разрешающей способностью, рабочей
областью спектра. Большая светосила необходима для регистрации слабых излучений,
большая дисперсия-для разделения спектральных линий с близкими длинами волн
при анализе в-в с многолинейчатыми спектрами, а также для повышения чувствительности
анализа. В качестве устройств, диспергирующих свет, используют дифракц. решетки
(плоские, вогнутые, нарезные, голографич., профилированные), имеющие от неск.
сотен до неск. тысяч штрихов на миллиметр, значительно реже-кварцевые или стеклянные
призмы.
Спектрографы (рис. 2),
регистрирующие спектры на спец. фотопластинках или (реже) на фотопленках , предпочтительнее
при качественном АЭСА, т. к. позволяют изучать сразу весь спектр образца (в
рабочей области прибора); однако используются и для количеств. анализа вследствие
сравнит. дешевизны, доступности и простоты обслуживания. Почернения спектральных
линий на фотопластинках измеряют с помощью микрофотометров (микроденситометров).
Использование при этом ЭВМ или микропроцессоров обеспечивает автоматич. режим
измерений, обработку их результатов и выдачу конечных результатов анализа.

Рис.2. Оптическая схема
спектрографа : 1-входная щель; 2-поворотное зеркало; 3-сферич. зеркало; 4-дифракц.
решетка; 5-лампочка освещения шкалы; 6-шкала; 7-фотопластинка.

Рис. 3. Схема квантометра
(из 40 каналов регистрации показано только три): 1-полихроматор; 2-дифракц.
решетки; 3-выходные щели; 4-ФЭУ; 5-входные щели; 6 - штативы с источниками света;
7 - генераторы искрового и дугового разрядов; 8- электронно-регистрирующее устройство;
9 - управляющий вычислит. комплекс.
В спектрометрах осуществляется фотоэлектрич. регистрация аналит. сигналов с помощью фотоэлектронных умножителей (ФЭУ) с автоматич. обработкой данных на ЭВМ. Фотоэлектрич. многоканальные (до 40 каналов и более) полихроматоры в квантометрах (рис. 3) позволяют одновременно регистрировать аналит. линии всех предусмотренных программой определяемых элементов. При использовании сканирующих монохроматоров многоэлементный
анализ обеспечивается высокой скоростью сканирования по спектру в соответствии с заданной программой.Для определения элементов
(С, S, P, As и др.), наиб, интенсивные аналит. линии к-рых расположены в УФ
области спектра при длинах волн меньше 180-200 нм, применяют вакуумные спектрометры.
При использовании квантометров
длительность анализа определяется в значит. мере процедурами подготовки исходного
в-ва к анализу. Существенное сокращение времени пробоподготовки достигается
автоматизацией наиб. длительных этапов - растворения , приведения р-ров к стандартному
составу, окисления металлов , растирания и смешения порошков , отбора проб заданной
массы. Во мн. случаях многоэлементный АЭСА выполняется в течение неск. минут,
напр.: при анализе р-ров с использованием автомати-зир. фотоэлектрич. спектрометров
с ВЧ плазматронами или при анализе металлов в процессе плавки с автоматич. подачей
проб в источник излучения.
В черной и цветной металлургии распространены экспрессные полуколичественные (относит. стандартное отклонение 0,3-0,5 и более) методики определения содержания основных или наиб. характерных компонентов сплавов , напр. при их маркировке, при сортировке металлолома для его утилизации и т.д. Для этого применяют простые, компактные и дешевые визуальные и фотоэлектрич. приборы (стило-скопы и стилометры) в сочетании с искровыми генераторами. Диапазон определяемых содержаний элементов-от неск. десятых долей процента до десятков процентов.

АЭСА применяют в научных
исследованиях; с его помощью открывали хим. элементы, исследуют археологич.
объекты, устанавливают состав небесных тел и т.д. АЭСА широко применяется также
для контроля технол. процессов (в частности, для установления состава исходного
сырья, технол. и готовых продуктов), исследования объектов окружающей среды
и др. С помощью АЭСА можно определять практически все элементы периодич. системы
в весьма широком диапазоне содержаний - от 10 -7 % (пкг/мл) до десятков
процентов (мг/мл). Достоинства АЭСА: возмож
ность
одновременного определения в малой навеске в-ва большого числа элементов (до
40 и более) с достаточно высокой точностью (см. табл.), универсальность методич.
приемов при анализе разл. в-в, экспрессность, сравнительная простота, доступность
и дешевизна аппаратуры.
, под
ред. Х.И. Зильберштейна, Л., 1987; Кузяков Ю.Я., Семененко К.А., Зо-ров Н.Б.,
Методы спектрального анализа, М., 1990. Ю.И. Коровин,
Спектральные методы исследования основаны на использовании явлений поглощения (или испускания) электромагнитного излучения атомами или молекулами определенного вещества.
Частота (длина волны) излучения определяется составом вещества. Интенсивность аналитического сигнала пропорциональна количеству частиц, вызвавших его появление, то есть массе определяемого вещества или компонента смеси.
Спектральные методы анализа дают широкие возможности для наблюдения и исследования соответствующих сигналов в различных областях электромагнитного спектра – рентгеновское и ультрафиолетовое излучение, видимый свет, инфракрасное, а также микро- и радиоволновое излучение.
По источнику и типу аналитического сигнала спектральные методы разделяют на молекулярно-абсорбционную спектрометрию, молекулярно-люминисцентную или флуориметрию, на атомно-абсорбционную и атомно-эмиссионную, а также спектрометрию ядерно-магнитного резонанса и электронно-парамагнитного резонанса.
В молекулярно-абсорбционной спектрометрии исследуют аналитические сигналы в области от 200 до 750 нм (УФ – излучение и видимый свет), вызванные электронными переходами внешних валентных электронов, а также поглощение излучения в ИК – и микроволновой области, связанное с изменением вращения и колебания молекул. Наиболее широкое распространение получил метод, основанный на изучении поглощения в видимой области спектра в интервале длин волн от 400 до 750 нм - фотометрия; а также метод, основанный на поглощении излучения в различных частях инфракрасной области электромагнитного спектра – ИК-спектрометрия, чаще всего используют поглощение излучения в средней (длина волны 2,5 - 25 мкм) и ближней (длина волны 0,8 - 2,5 мкм) ИК-области.
Фотометрический метод количественного анализа основан на способности определяемого вещества, компонента смеси или их окрашенных форм поглощать электромагнитное излучение оптического диапазона. Способность к поглощению зависит от цветности исследуемого вещества. Цветность определяется электронным строением молекулы, обычно ее связывают с наличием в молекуле так называемых хромофорных групп, обусловливающих поглощение электромагнитного излучения веществом в видимой и УФ-областях спектра.
Общая схема выполнения фотометрического определения едина и включает следующие стадии:
· подготовку пробы и переведение определяемого вещества или компонента в раствор, в реакционноспособную, в зависимости от химизма аналитической реакции, форму;
· получение окрашенной аналитической формы определяемого вещества в результате проведения цветной реакции при оптимальных условиях, обеспечивающих ее избирательность и чувствительность;
· измерение светопоглощающей способности аналитической формы, т. е. регистрация аналитического сигнала при определенных условиях, отвечающих его локализации и наибольшей интенсивности.
Промышленностью выпускаются различные приборы молекулярно-абсорбционной спектрометрии - колориметры, фотометры, фотоэлектроколориметры, спектрофотометры и т.д., в которых установлены различные комбинации источников света, монохроматизаторов и рецепторов. Приборы можно классифицировать следующим образом:
· по способу монохроматизации лучистого потока - спектрофотометры, т. е. приборы с призменным или решеточным монохроматором, позволяющие достигать высокой степени монохроматизации рабочего излучения; фотоэлектроколориметры, т. е. приборы, в которых монохроматизация достигается с помощью светофильтров;
· по способу измерения - однолучевые с прямой схемой измерения (прямопоказывающие) и двухлучевые с компенсационной схемой;
· по способу регистрации измерений - регистрирующие и нерегистрирующие.
Инфракрасная спектроскопия – это метод анализа химических соединений, при котором поглощается энергия в пределах инфракрасного излучения (тепловое излучение). ИК-спектроскопию применяют для определения практически любой функциональной группы, идентификации соединений и т.п. Различные молекулы, содержащие одну и ту же атомную группировку, дают в ИК-спктре полосы поглощения в области одной и той же характеристической частоты. Характеристические частоты дают возможность по спектру установить конкретные группы атомов в молекуле и тем самым определить качественный состав вещества и строение молекулы. В общем случае ИК – анализатор представляет собой однолучевой или двухлучевой инфракрасный спектрофотомер, состоящий из трех основных блоков: подготовки пробы, спектрофотометрических измерений, преобразования сигналов и вычислений. В настоящее время разработаны ИК-анализаторы, основанные на работе в ближайшей инфракрасной области спектра 0,8-2,5 мкм (БИК-анализаторы).
Молекулярно-люминисцентная спектрометрия. Метод анализа, основанный на измерении флуоресценции, называется флуориметрией . Флуоресценция (люминисценция – испускание света) обусловлена поглощением веществом света определенной длины волны. Поглощение ультрафиолетового света определенными молекулами с легковозбуждаемыми электронами приводит к флуоресценции в видимой спектральной области. Флуоресценция свойственна относительно небольшому числу соединений (ароматическим соединениям и порфинам). Ряд соединений можно перевести во флуоресцирующие, введя в молекулу флуоресцирующую группу. Основным преимуществом флуориметрии по сравнению с другими абсорбционными методами является высокая селективность, так как флуоресценцией обладает значительно меньшее число веществ. Метод применяют для чувствительного определения очень малых количеств элементов при анализе органических веществ, при определении малых количеств витаминов, гормонов, антибиотиков, канцерогенных соединений и др. Флуориметрию используют для определения микроорганизмов и соматических клеток.
Методика определения микроорганизмов состоит в специальной подготовке пробы, в процессе которой бактерии, содержащиеся в продукте, окрашиваются красителем в ярко-оранжевый цвет, в результате чего бактериальная суспензия приобретает способность флуоресцировать. Интенсивность флуоресценции пропорциональна числу микробов и контролируется электронным способом.
Флуориметрический метод контроля микроорганизмов достаточно универсален, имеет несложное аппаратурное оформление.
В атомной спектроскопии вещества исследуют, переведя их в состояние атомного пара – атомно-абсорбционная спектроскопия или газообразное состояние – атомно-эмиссионная спектроскопия. Метод атомной спектроскопии находит широкое применение при анализе различных видов сырья и пищевых продуктов. Метод позволяет определить около 70 различных элементов; используется для одновременного определения большого числа элементов (многоэлементарный анализ); для серийного анализа, благодаря высокой чувствительности и быстроте.
Атомно-абсорбционная спектрометрия основана на измерении поглощения электромагнитного излучения атомным паром анализируемого вещества. Измеряют фотометрически разность интенсивности излучения до и после прохождения через анализируемый образец. Прибором, позволяющим осуществить метод ААС, является
· атомно-абсорбционный спектрометр, имеющий следующие основные составные части,
· источник света определенной длины волны, характерной для исследуемого металла;
· «абсорбционную ячейку», в которой происходит атомизация пробы;
· монохроматор для выделения узкой части спектра определенной длины волны;
· фотоумножитель, который детектирует, усиливает и измеряет интенсивность результирующего светового потока;
· регистрирующее и записывающее результирующий сигнал устройство.
Источник света испускает поток луча, спектр которого характерен для определяемого элемента. Этот поток фокусируется через абсорбционную ячейку и монохроматор, где выделяется характерная для исследуемого элемента область спектра. Затем поток направляется в фотоумножитель и преобразуется в электрический сигнал. Величина последнего зависит от интенсивности поступающего в фотоумножитель светового потока и регистрируется специальным устройством.
Сравнивая результаты измерений в исследуемой пробе с результатами измерений в стандартных растворах, определяют содержание элемента в пробе.
В качестве источников излучения при определении содержания рассматриваемых металлов, как правило, используют лампы с полым катодом, являющиеся источниками линейных спектров. Катод такой лампы имеет форму полого цилиндра или стакана. Объем лампы заполнен инертным газом (неоном или иногда аргоном). Существуют многоэлементные лампы с полым катодом, например для определения содержания меди и цинка или меди, цинка, свинца и кадмия. Они бывают очень удобны. Их преимуществом является снижение затрат времени на прогрев ламп. Однако такие лампы, как правило, продуцируют излучение более низкой энергии, чем одноэлементные лампы, что в результате приводит к ухудшению чувствительности; могут возникать спектральные помехи.
В атомно-эмиссионной спектрометрии исследуют атомно-эмиссионные спектры, полученные в результате возбуждения атомов в газообразном состоянии.
Для перевода атомов в газообразное состояние и их возбуждения используют плазму, в качестве среды для получения плазмы применяют аргон. Существует два способа получения плазмы. В одном из них возбуждение происходит под действием электрических разрядов между электродами - плазма постоянного тока, а в другом- энергия высокочастотного переменного тока передается газу с по- мощью магнитной индукции - индуктивно-связанная плазма. При этом создаются высокие температуры, благодаря которым большинство атомов переходит в возбужденное состояние. Поглощение энергии такими атомами невозможно, поэтому при переходе из возбужденного состояния в основное происходит эмиссия (испускание) фотонов, интенсивность которой пропорциональна числу возбужденных атомов. Количественное определение элемента производят так же, как в атомно-абсорбционной спектрометрии.
Приборы, позволяющие осуществить метод АЭС, имеют те же основные части, что и атомно-абсорбционный спектрометр.
Спектроскопия магнитного резонанса. Масс-спектроскопия . Спектроскопия ядерного магнитного резонанса (ЯМР)изучает магнитный резонанс, возникающий в результате взаимодействия магнитного момента ядра с внешним магнитным полем. С помощью метода ЯМР можно исследовать ядра с собственным момент количества движения (спин ядра) и связанным с ним магнитным моментом ядра.
Согласно квантовой механике собственный момент количества движения (спин) ядра принимает строго определенные значения. Так как спин ядра является вектором, то он характеризуется величиной и направлением. Во внешнем магнитном поле для спина ядра возможны две ориентации: вдоль и против направления силовых линий внешнего магнитного поля. Каждому значению спина соответствует определенное значение энергии. Переориентация спина ядра с изменением направления сопровождается поглощением энергии DЕ. Такие переходы вызываются воздействием на ядро радиочастотной области электромагнитного спектра. При этом анализируемая система поглощает энергию при строго фиксированных значениях частоты v, т. е. наблюдается явление резонанса. Такое поглощение энергии измеряют экспериментально, DE прямо пропорционально напряженности магнитного поля в месте расположения ядра и определяется как DЕ= hv, где h – постоянная Планка.
Спектроскопия электронного паромагнитного резонанса (ЭПР) изучает магнитный резонанс, возникающий в результате взаимодействия магнитного момента электрона с внешним высокочастотным (микроволновым) магнитным полем. Метод ЭПР служит для исследования внутримолекулярного окружения неспаренных электронов.
Теория магнитного резонанса применима не только к ядрам, но и к электронам, поскольку последние также имеют спин и магнитный момент. В отсутствие внешнего магнитного поля спины электронов беспорядочно ориентированы, а энергия электронов одинакова. В постоянном магнитном поле магнитные моменты электронов ориентированы соответственно направлению внешнего магнитного поля. Электроны с ориентацией спинов вдоль поля находятся на высоком энергетическом уровне, электроны с ориентацией против поля - на низком, более стабильном, уровне. Если на электроны, находящиеся в однородном магнитном поле воздействовать высокочастотным магнитным полем, направление, которого перпендикулярно направлению однородного магнитного поля, то при определенных соотношениях между напряженностью постоянного поля и частотой переменного поля наблюдается резонансное поглощение энергии переменного поля. Оно регистрируется на спектрометре в виде спектра электронного парамагнитного резонанса - ЭПР-спектра. При количественной оценке спектра в качестве основного аналитического параметра используют константу спин-спинового взаимодействия Масс-спектроскопия занимает особое положение среди спектроскопических методов. В строгом смысле слова этот метод не является спектроскопическим, так как вещество при анализе не подвергается воздействию электромагнитного излучения.
Масс-спектроскопия основана на изучении тока от фрагментов ионов, полученных из нейтральных молекул вещества путем воздействия на них пучка электронов.
Вещество, исследуемое методом ядерно-магнитного резонанса , помещают одновременно в два магнитных поля - одно постоянное, а другое радиочастотное. Измерение осуществляют на ЯМР-спектрометре, основными составляющими элементами которого являются: электромагнит (в простых приборах используют постоянный магнит); генератор радиочастотного излучения; датчик, в который помещают пробирку с образцом, электронный усилитель и интегратор, самописец. В ЯМР-методе используют следующие аналитические параметры: химический сдвиг, константа спин-спинового взаимодействия, интенсивность сигнала, время релаксации.
Метод электронного парамагнитного резонанса основан на измерении поглощения веществом энергии внешнего магнитного поля. Метод ЭПР применяют для анализа всех соединений, содержащих неспаренные электроны, независимо от их агрегатного состояния. Область применения определяется конструкцией кюветы. ЭПР является одним из самых чувствительных методов, предел чувствительности составляет10 "моль/л.
Масс-спектрометрия основана на получении ионов из нейтральных молекул путем воздействия на них пучком электронов, обладающих энергией, достаточной для ионизации. При этом, главным образом, образуются положительные ионы, которые могут распадаться на отдельные фрагменты. Регистрируемая завис мость ионных токов от массы отдельных фрагментов называется масс-спектром. Молекула, возбужденная в результате взаимодействия с электроном (с энергией более 10з кДж/моль), распадается с образованием положительного молекулярного иона и электрона (ионизация).
В большинстве случаев молекулярный ион обладает значительной внутренней энергией и быстро распадается далее с образованием заряженных и незаряженных фрагментов (фрагментация).
Осколочные ионы, в свою очередь, могут распадаться с образованием новых фрагментов. В некоторых случаях фрагментация сопровождается перегруппировками. Процесс фрагментации молекулярных ионов происходит до тех пор, пока не образуют ионы, внутренней энергии которых недостаточно для их дальнейшего превращения. Масс-спектрометры работают при высоком вакууме, что сводит к минимуму нежелательные межмолекулярные реакции и, кроме того, благоприятствует внутримолекулярной фрагментации.
Масс-спектр представляет собой спектр линий положительно заряженных ионов. Несмотря на то что реальной связи между масс-спектрометрией и оптической спектрометрией не существует, оба метода называют спектрометрическими из-за формального сходства графических изображений спектров.
Метод масс- спектрометрии применяют в научно-исследовательской практике для идентификации соединений и установления строения неизвестных веществ, точного определения молекулярной массы, определения элементарного состава, анализа следовых количеств биологически активных соединений, определения аминокислотной последовательности пептидов, анализа многокомпонентных смесей и т.п.
Macс -спектральный анализ основан на способности газообразных ионов разделяться в магнитном поле в зависимости от отношения m/е, где m - масса, е - заряд иона. Ионизация молекул в газе происходит под действием потока электронов. По величине m/е определяют массовое число иона, а по интенсивности соответствующего сигнала судят о концентрации ионов.
Качественный масс-спектральный анализ основан на измерении массы ионов. Идентификация масс поводится по положению линии на фотопластинке, которое фиксируют, измеряя расстояние между линиями с известной массой и анализируемой линией.
Количественные измерения в масс -спектрометрии проводят по току, фиксируемому детектором, или по почернению фотопластинки. В первом случае расчеты основаны на том, что пик ионного тока I пропорционален содержанию компонента или его парциальному давлению:
Где k, c - коэффициенты пропорциональности; с - концентрация; р - давление.
Спектральные методы анализа основаны на изучении оптических спектров испускания или поглощения. Различают атомно-абсорбционный метод спектрального анализа (анализ по спектрам поглощения) и эмиссионный спектральный анализ (анализ по спектрам испускания). Спектральный анализ широко применяют для качественного и количественного анализа различных веществ. По характеристическим линиям спектра можно определять элементный состав вещества, а интенсивность спектральной линии является мерой концентрации вещества в пробе.
Атомы элементов в возбужденном состоянии испускают излучение со строго определенной длиной волны. Спектры испускания (эмиссионные спектры) для каждого элемента индивидуальны, они состоят из определенного набора характерных линий, по которым можно определять элементный состав вещества и его концентрацию.
При эмиссионном спектральном анализе исследуемую пробу испаряют или сжигают, если это жидкое или твердое вещество, затем подвергают действию высокой температуры или электрического заряда для перевода атомов в возбужденное состояние и регистрируют спектр. Качественный эмиссионный анализ сводится к расшифровке линий в спектре анализируемого образца. Количественный анализ основан на сравнении интенсивности спектральных линий образца с интенсивностью линий в спектре стандартного образца, содержание определяемого элемента в котором известно.
Источниками возбуждения могут служить пламя, электрическая дуга, искра, импульсный или электровакуумный разряд. Дуговой разряд дает температуру 5000-7000 °С, при которой в возбужденное состояние переходят атомы большинства элементов. В высоковольтной искре с температурой 7000-15000 °С возбуждаются атомы элементов с высоким потенциалом возбуждения. Импульсный и электровакуумные разряды используют для возбуждения инертных газов.
По методу регистрации спектра различают несколько видов эмиссионного спектрального анализа. При визуальном анализе качественный состав определяют непосредственным наблюдением видимого спектра. Более точен фотографический анализ, по которому спектр фотографируют на фотопластинку, которую затем рассматривают на спектропроекторе при качественных определениях или фотометрируют с помощью микрофотометра при количественных определениях. На фотографической пластинке получают фиксированный ряд линий, соответствующих спектральным линиям исследуемого образца, степень почернения которых пропорциональна интенсивности этих линий.
Для расшифровки спектрограмм используют спектропроекторы. Отечественной промышленностью выпускается спектропроектор ПС-18, который дает возможность получить на экране увеличенные в 20 раз небольшие участки спектра, облегчая их расшифровку при экспрессном качественном или полуколичественном анализе.
Плотность почернения линий на фотопластинке измеряют с помощью микрофотометров. Световой поток пропускают через незачерненную часть фотопластинки, а затем направляют его на фотоэлемент с гальванометром. Отмечают отклонение стрелки гальванометра по шкале. Затем световой поток пропускают через зачерненную часть пластинки и снова отмечают отклонение стрелки гальванометра. Плотность почернения определяют по уравнению:
где I0 - интенсивность света, прошедшего через незачерненную часть фотопластинки; I - интенсивность света, прошедшего через зачерненную часть фотопластинки.
Поскольку плотность почернения пропорциональна концентрации элемента, по показаниям гальванометра строят градуировочный график зависимости почернения от концентрации. По такому графику затем определяют содержание элемента. Для определения плотности почернения линий на спектрограмме применяют микрофотометр МФ-2 (или МФ-4) и двухлучевой микрофотометр ИФО-451.
При фотоэлектрическом эмиссионном анализе аналитические линии регистрируют с помощью фотоэлементов. Результат анализа указывается на шкале измерительного прибора или фиксируется на ленте самозаписывающего прибора.
Кварцевый спектрограф ИСП-28. Спектрограф ИСП-28 используют для получения спектров в интервале длин волн 200-600 нм. На нем проводят качественный и количественный анализы металлов, сплавов, руд, минералов и других материалов. На рис. 126 показана оптическая схема прибора. Свет от источника 1 (дуга или искра) через трехлинзовый конденсор 3-5, защищенный от брызг металлов кварцевой пластинкой 2, направляется в щель 6, находящуюся в фокусе зеркального объектива 8. Отраженный от этого объектива параллельный пучок света направляется на кварцевую призму 9. Подвергшийся дисперсии свет кварцевым объективом 10 фокусируется на эмульсии фотопластинки 11.

Другие спектрографы. Кварцевый лабораторный спектрограф ИСП-30 настольного типа применяется для качественного анализа металлов, сплавов и руд; стеклянный трехпризменный спектрограф ИСП-51 используется для анализа веществ, содержащих элементы с малым числом спектральных линий. Для анализа веществ, содержащих элементы с особо сложными спектрами, используют спектрограф СТЭ-1. Для качественного и количественного анализа металлов, руд, минералов и др. применяют длиннофокусный спектрограф ДФС-8 (три модификации) с дифракционными решетками и дифракционный спектрограф ДФС-452.
Пламенная фотометрия является одним из наиболее точных методов эмиссионного спектрального анализа. Этот метод широко применяют для определения щелочных и щелочноземельных металлов. Сущность метода пламенной фотометрии заключается в следующем.
Раствор анализируемого вещества сжатым воздухом разбрызгивается в зону пламени газовой горелки, в которой сгорают ацетилен, водород, светильный или какой-либо другой газ. Пламя горелки служит также источником энергии для возбуждения атомов. Оптическое устройство выделяет спектральную линию определяемого элемента и измеряет ее интенсивность с помощью фотоэлемента. Интенсивность спектральной линии пропорциональна концентрации соли в растворе (в определенных границах). Концентрацию элемента определяют по градуировочному графику. Ниже приведены состав некоторых горючих газовых смесей и средняя температура, получаемая при их сжигании (в °С):

Портативный пламенный фотометр ППФ-УНИЗ. Принципиальная схема фотометра ППФ-УНИЗ представлена на рис. 127. Горючий газ из баллона (или городской сети) проходит через маностат 2, буферную бутыль 3, фильтр 4 и поступает через микрокран 5 в смеситель 7, выполняющий одновременно функцию каплеуловителя. Давление газа после маностата поддерживается постоянным с помощью микрокрана 5 и измеряется U-образным жидкостным манометром 6. Избыток газа выходит в лабораторную горелку 1 и сжигается.

Сжатый воздух из компрессора (без применения масляной смазки) или из баллона поступает в буферную бутыль 3", затем в фильтр 13. Давление воздуха поддерживается постоянным с помощью микрокрана 12 и измеряется манометром 11. Воздух поступает в распылитель 8, куда засасывается анализируемый раствор из стакана 10. Раствор в виде мелкораспыленного аэрозоля поступает в смеситель 7, где смешивается с горючим газом. Выходящая из смесителя газовоздушная смесь, содержащая в распыленном состоянии исследуемый элемент, через каплеуловитель 14 поступает в горелку 20.
Длина волны желтой линии пламени натрия составляет 589±5 мкм, красной линии кальция - 615±5 мкм, инфракрасной линии калия - 766±5 мкм. Интенсивность этих линий фиксируют фотоэлементом 16, снабженным сменными интерференционными светофильтрами 17 и диафрагмами 18. При определении натрия и кальция используют селеновые фотоэлементы типа АФИ-5 с чувствительностью 460-500 мкА/лм, для определения калия - сернисто-серебряный фотоэлемент типа ФЭСС-УЗ с чувствительностью 6000-9000 мкА/лм. Фотоэлементы и светофильтры защищены от прямого теплового излучения пламени стеклянным экраном 19. Возникающие фототоки регистрируются магнитоэлектрическим микроамперметром 21 типа М-95, к которому два из трех фотоэлементов присоединены по компенсационной схеме через электрический переключатель 15.
Перед началом работы с прибором открывают дверку 10 (рис. 128) и закрепляют ее с помощью фиксатора. К сливной трубке 14 распылителя 12 подсоединяют резиновую трубку и опускают ее в сосуд с запорной жидкостью высотой 20-25 см. Под всасывающую трубку 13 распылителя подставляют стакан вместимостью 25-30 мл с дистиллированной водой. На дверку устанавливают защитное устройство (козырек) 11 и включают прибор в сеть переменного тока в 220 В (50 Гц). Включают компрессор для подачи воздуха и, медленно вращая рукоятку микрокрана «воздух» 4 против часовой стрелки, добиваются хорошего распыления дистиллированной воды, т.е. образования высокодисперсного аэрозоля. Оптимальное давление воздуха (4-8)*10000 Па (0,4-0,8 атм) не должно изменяться в течение всего времени измерения.

Медленно вращая рукоятку микрокрана «газ» 5, подают газ в горелку и через 10-20 с зажигают его у входа в горелку и на выходе из маностата. Подачу газа регулируют так, чтобы внутренний конус пламени окрашивался в зеленый цвет, а внешний - в голубовато-синий. С помощью рукоятки 9 устанавливают горелку в таком положении, при котором внутренний конус пламени опущен на 5-6 см ниже кромки входного отверстия диафрагмы.
Измерения начинают после 20-минутного прогревания фотометрической ячейки. В период прогревания диафрагма ячейки должна быть полностью открыта, микроамперметр включают на низкую чувствительность (1,0 мкА) и в пламя горелки вводят дистиллированную воду. После прогревания фотоэлектрической ячейки диафрагму закрывают, рукоятку микроамперметра 6 переключают на высшую чувствительность (0,1 мкА) и указатель микроамперметра устанавливают на нуль, вращая головку корректора, находящуюся на правой боковой стороне прибора.
Для построения градуировочного графика готовят серию стандартных растворов. Для приготовления исходного раствора 2,385 г хлорида калия KCl (хч) растворяют в мерной колбе вместимостью 500 мл и разбавляют водой до метки. Отбирают пипеткой 5,00 мл этого раствора в мерную колбу вместимостью 500 мл и разбавляют дистиллированной водой до метки (разбавление в 100 раз). Полученный раствор содержит 25 мг калия в 1 мл, из него готовят растворы, содержащие 5, 10, 15 и 20 мг калия в 1 мл. Для этого в мерные колбы вместимостью 100 мл отбирают пипеткой 20, 40, 60 и 80 мл раствора с содержанием калия 25 мг/мл и разбавляют объем водой до метки.
Эти растворы последовательно вводят в пламя горелки и записывают показания микроамперметра. При переходе от одного раствора к другому распылитель промывают дистиллированной водой до возвращения стрелки микроамперметра к нулю. По полученным данным строят градуировочный график: показания микроамперметра (по оси абсцисс) - концентрация определяемого элемента (по оси ординат) (в мг/мл).
Для определения концентрации элемента в исследуемом растворе его вводят в пламя горелки и записывают показания микроамперметра, по которым, пользуясь градуировочным графиком, находят концентрацию определяемого элемента. В течение всего процесса анализа необходимо поддерживать постоянство давления воздуха и газа.
Кроме метода определения концентрации по градуировочному графику применяют метод ограничивающих растворов, т.е. снимают показания микроамперметра при анализе исследуемого раствора и параллельно показания прибора при анализе стандартных: растворов с меньшей и большей концентрацией. Содержание калия (в мг/л) вычисляют по формуле
![]()
где c1 - содержание калия в более концентрированном стандартном растворе; c2 - содержание калия в менее концентрированном стандартном растворе; I1 - показания микроамперметра при анализе стандартного раствора с большей концентрацией; I2 - показания микроамперметра при анализе стандартного раствора с меньшей концентрацией; Ix - показания микроамперметра при анализе исследуемого раствора.
Пламенный фотометр Flapho-4. Двухканальный прибор для серийного определения содержания натрия, калия, кальция, лития и свинца с высокой чувствительностью. Выпускается в ГДР.
Исследуемый раствор пробы всасывается протекающим через; распылитель сжатым воздухом и превращается в аэрозоль. Аэрозоль поступает в специальный резервуар, где к нему примешивается горючий газ (ацетилен или пропан), и полученная смесь подводится к горелке, окруженной очищенным воздухом. В газовом пламени исследуемое вещество испаряется, и его атомы возбуждаются. Металлизированный интерференционный фильтр выделяет из общего спектра пламени монохроматический компонент излучения, который попадает на селеновый фотоэлемент. Образующийся прерывистый фототок усиливается и подводится к измерительному или регистрирующему прибору. Схема прибора представлена на рис. 129.

Другие пламенные фотометры: фотометр пламенный ФП-101 трехканальный для определения концентрации Na, K, Ca и Li; фотометр пламенный ПФМ для количественного определения концентраций щелочных и щелочноземельных элементов, а также магния, бора, хрома и марганца; пламенно-фотометрические анализаторы жидкости ПАЖ-1 и БИАН-140 для определения микроколичеств K, Na, Ca и Li в растворах, фотометр пламенный для определения Na и K в биологических жидкостях.
Свободные атомы в невозбужденном состоянии, находящиеся в зоне низкотемпературного пламени, обладают способностью избирательно поглощать свет. Длина волны света, поглощаемого атомами элемента, совпадает с длиной волны света, испускаемого атомами этого элемента. Следовательно, по характеристическим линиям спектра поглощения и их интенсивности можно проводить анализ веществ, определяя их состав и концентрацию составляющих его элементов.
Для проведения атомно-абсорбционного анализа исследуемое вещество испаряют, подавая его в зону низкотемпературного пламени. Молекулы испарившегося вещества диссоциируют на атомы. Поток света, в спектре которого имеется линия света, поглощаемая веществом, пройдя через это пламя, ослабляется, и тем больше, чем выше концентрация анализируемого вещества.
На рис. 130 представлена принципиальная схема установки для атомно-абсорбционного анализа. Свет от разрядной трубки 1 (полый катод) проходит через пламя горелки 2 и фокусируется на щели монохроматора 3. Затем излучение попадает на фотоумножитель, или фотоэлемент 4. Монохроматор выделяет из общего светового потока излучение с длиной волны, поглощаемой исследуемым элементом. Ток усиливается в блоке 5 и регистрируется измерительным устройством 6.

Определение заключается в измерении отношения интенсивностей света, прошедшего через пламя с введенным в него анализируемым веществом и без него. Поскольку интенсивность спектральной линии исследуемого элемента в пламени горелки оказывается больше, чем их интенсивность излучения от полого катода, излучение последнего модулируют. Модуляция излучения (изменение амплитуды и частоты колебаний) осуществляется с помощью вращающегося диска с отверстиями (модулятор 7), расположенного между полым катодом и пламенем. Усилитель 5 должен иметь максимальный коэффициент усиления для той же частоты, с которой модулируется излучение полого катода.
Атомно-абсорбционный спектрофотометр AAS-1. Предназначается для абсорбционного и эмиссионного спектрального анализа. Дает возможность определять 65 элементов.
Принцип действия. Жидкая проба распыляется с помощью газа-окислителя, смешивается с горючим газом (ацетилен или пропан) и сжигается в пламени горелки. Через пламя горелки проходит излучение от лампы с полым катодом. После выделения дифракционным монохроматором подходящей линии излучение направляется на фотоумножитель. Постоянная составляющая тока, вызванная собственным излучением, подавляется. Сигнал от фото-умножителя усиливается, выпрямляется чувствительным выпрямителем и регистрируется. Прибор настраивается и контролируется по стандартным растворам.
На рис. 131 приведена схема атомно-абсорбционного спектрофотометра AAS-1.

Устройство прибора. Прибор имеет арматурный комплекс для снабжения газами, систему распыления и сжигания, сменное устройство для ламп с полыми катодами, оптическую систему я приемное устройство с усилителем и индикатором.
Пламя горелки питается смесью ацетилена или пропана и сжатого воздуха. Газы поступают в систему сжигания из обычных баллонов с отрегулированными (первичными) редукторами давления. Подача воздуха, свободного от масла, обеспечивается мембранным компрессором (16 л/мин под давлением 3*100000 Па (3 атм)). Арматурный комплекс прибора имеет регулируемые (вторичные) редукторы и расходомеры для контроля расхода каждого газа, а также керамические спеченные пылевые фильтры и склянку для дополнительного промывания ацетилена. Предохранительный клапан автоматически прекращает доступ горючего газа при снижении рабочего давления сжатого воздуха (например, вследствие перегиба или отрыва подводящего шланга); клапан исключает неправильный порядок подачи газов при зажигании пламени.
Система распыления и сжигания находится за съемным окном из многослойного стекла, позволяющего наблюдать за работой системы. Распылитель с кольцевым соплом обладает большим коэффициентом распыления и характеризуется низким расходом жидкости (3,4 мл/мин, или 0,5 мл за время всего анализа). Горелка оснащена сменными головками-насадками - одной щелевой для абсорбционного анализа (рис. 132, а) и двумя многодырчатыми (горелками Мекера с сеткой) для эмиссионного анализа (рис. 132,6).

Юстируемые держатели для четырех ламп с полыми катодами находятся в устройстве, позволяющем осуществлять быструю смену ламп. После замены одной из ламп держатели в юстировке не нуждаются.
Оптическая система направляет излучение лампы в виде узкого пучка на пламя. За счет бокового смещения тубуса с изображающей системой добиваются однократного или трехкратного прохождения излучения через пламя для повышения чувствительности анализа. Светосильный дифракционный монохроматор выделяет из линейчатого спектра данной лампы с полым катодом желаемую резонансную линию. Ширину щели монохроматора регулируют в пределах от 0 до 2 мм.
Прецизионная дифракционная решетка с 1300 штрихами на 1 мм и угловой дисперсией 1,5 нм/мм обладает большой разрешающей способностью. Спектральный интервал решетки от 190 до 820 нм.
Приемником излучения служит 12-каскадный фотоумножитель. Измерительный усилитель, блок питания ламп с полым катодом и фотоумножители работают на транзисторах и способны компенсировать колебания напряжения сети от +10 до -15%.
Показания прибора отсчитывают по стрелочному индикатору, имеющему три шкалы: логарифмическая шкала коэффициента погашения от 0 до 1,5; линейная шкала от 0 до 100 и шкала рабочих напряжений от 0 до 16 мВ. К прибору может быть подключено регистрирующее или вычислительное устройство для определения концентрации или для обработки данных. Чувствительность определений (в мг/л) составляет:
![]()
Прибор работает от сети переменного тока 220 В, 50 Гц. Выпускается в ГДР.
Другие отечественные атомно-абсорбционные спектрофотометры: атомно-абсорбционный спектрофотометр С-302 для определения микроколичеств железа, меди, цинка, кобальта, никеля, висмута, кальция и других элементов; автоматизированный атомно-абсорбционный спектрофотометр АА-А для определения кальция и меди с повышенной чувствительностью; «Сатурн» - пламенный атомно-абсорбционный полуавтоматический регистрирующий спектрофотометр для определения 32 элементов; «Спектр-1» - атомно-абсорбционный спектрофотометр для экспрессного определения более 40 элементов чувствительностью примерно 0,2 мкг/мл.
В Англии выпускается атомно-абсорбционный спектрофотометр Перкин-Эльмер, модель 603. Прибор построен по двухлучевой схеме, скомбинирован с микрокомпьютером. Обеспечивает высокую точность и экспрессность определения. Для зажигания пламени используется горючая смесь кислород-ацетилен.
Спектры излучения . Спектральный состав излучения у различных веществ имеет весьма разнообразный характер. Однако все спектры делятся на три типа: а) сплошной спектр; б) линейчатый спектр; в) полосатый спектр.
а) Сплошной (непрерывный) спектр . Накаленные твердые и жидкие тела и газы (при большом давлении) испускают свет, разложение которого дает сплошной спектр, в котором спектральные цвета непрерывно переходят один в другой. Характер непрерывного спектра и сам факт его существования определяются не только свойствами отдельных излучающих атомов, но и взаимодействием атомов друг с другом. Сплошные спектры одинаковы для разных веществ, и поэтому их нельзя использовать для определения состава вещества.
б) Линейчатый (атомный) спектр . Возбужденные атомы разреженных газов или паров испускают свет, разложение которого дает линейчатый спектр,состоящий из отдельных цветных линий. Каждый химический элемент имеет характерный для него линейчатый спектр. Атомы таких веществ не взаимодействуют друг с другом и излучают свет только определенных длин волн. Изолированные атомы данного химического элемента излучают строго определенные длины волн. Это позволяет по спектральным линиям судить о химическом составе источника света.
в) Молекулярный (полосатый) спектр .Спектр молекулы состоит из большого числа отдельных линий, сливающихся в полосы, четкие с одного края и размытые с другого. В отличие от линейчатых спектров полосатые спектры создаются не атомами, а молекулами, не связанными или слабо связанными друг с другом. Серии очень близких линий группируются на отдельных участках спектра и заполняют целые полосы. В 1860 г. немецкие ученые Г. Кирхгоф и Р. Бунзен, изучая спектры металлов, установили следующие факты:
1) каждый металл имеет свой спектр;
2) спектр каждого металла строго постоянен;
3) введение в пламя горелки любой соли одного и того же металла всегда приводит к появлению одинакового спектра;
4) при внесении в пламя смеси солей нескольких металлов в спектре одновременно появляются все их линии;
5) яркость спектральных линий зависит от концентрации элемента в данном веществе.
Спектры поглощения. Если белый свет от источника, дающей сплошной спектр, пропускается через пары исследуемого вещества и затем разлагается в спектр, то на фоне сплошного спектра наблюдаются темные линии поглощения в тех же самых местах, где находились бы линии спектра испускания паров исследуемого элемента. Такие спектры получили название атомных спектров поглощения.
Все вещества, атомы которых находятся в возбужденном состоянии, излучают световые волны, энергия которых определенным образом распределена по длинам волн. Поглощение света веществом также зависит от длины волны. Атомы поглощают излучение лишь тех длин волн, которые они могут испускать при данной температуре.
Спектральный анализ. Явление дисперсии используется в науки и технике в виде метода определения состава вещества, получившего название спектрального анализа. В основе этого метода лежит изучение света, излучаемого или поглощаемого веществом. Спектральным анализом называется метод изучения химического состава вещества, основанный на исследовании его спектров.
Спектральные аппараты . Для получения и исследования спектров используют спектральные аппараты. Наиболее простые спектральные приборы - призма и дифракционная решетка. Более точные - спектроскоп и спектрограф.
Спектроскопом называется прибор, с помощью которого визуально исследуется спектральный состав света, испускаемого некоторым источником. Если регистрация спектра происходит на фотопластинке, то прибор называется спектрографом.
Применение спектрального анализа . Линейчатые спектры играют особо важную роль, потому что их структура прямо связана со строением атома. Ведь эти спектры создаются атомами, не испытывающими внешних воздействий. Состав сложных, главным образом органических смесей анализируется по их молекулярным спектрам.
С помощью спектрального анализа можно обнаружить данный элемент в составе сложного вещества, если даже его масса не превышает 10 -10 г. Линии, присущие данному элементу, позволяют качественно судить о его наличии. Яркость линий дает возможность (при соблюдении стандартных условий возбуждения) количественно судить о наличии того или иного элемента.
Спектральный анализ можно проводить и по спектрам поглощения. В астрофизике по спектрам можно определить многие физические характеристики объектов: температуру, давление, скорость движения, магнитную индукцию и др. с помощью спектрального анализа определяют химический состав руд и минералов.
Основные направления применения спектрального анализа таковы: физико-химические исследования; машиностроение, металлургия; атомная индустрия; астрономия, астрофизика; криминалистика.
Современные технологии создания новейших строительных материалов (металлопластиковые, пластиковые) непосредственно взаимосвязаны с такими фундаментальными науками как химия, физика. Данные науки используют современные методы исследования веществ. Поэтому спектральный анализ можно применять для определения химического состав состава строительных материалов по их спектрам.
СПЕКТРАЛЬНЫЙ АНАЛИЗ (при помощи спектров испускания) имеет применение почти во всех отраслях хозяйства. Широко применяется в металлопромышленности для быстрого анализа железа, стали, чугуна, а также различных специальных сталей и готовых металлических изделий, для установления чистоты легких, цветных и драгоценных металлов. Большое применение имеет спектральный анализ в геохимии при изучении состава полезных ископаемых. В химической промышленности и близких к ней отраслях спектральный анализ служит для установления чистоты выпускаемой и применяемой продукции, для анализа катализаторов, различных остатков, осадков, мутей и промывных вод; в медицине - для открытия металлов в различных органических тканях. Ряд специальных задач, трудно разрешаемых или вовсе не разрешимых иным путем, решается при помощи спектрального анализа быстро и точно. Сюда относится, например, распределение металлов в сплавах, исследование в сплавах и минералах сульфидных и других включений; такого рода исследования иногда обозначаются термином локальный анализ .
Выбор того или другого типа спектрального аппарата с точки зрения достаточности его дисперсии производится в зависимости от цели и задач спектрального анализа. Для исследования платиновых металлов (Ru, Rh, Pd, Os, Ir, Pt), а также Fe, Co, Ni, Сг, V, Mo, W, Ti, Mn, Zr, Re, Nb и Та наиболее пригодны кварцевые спектрографы с большей дисперсией, дающие для длин волн 4000-2200 Ӑ полоску спектра длиной по крайней мере 22 см. Для остальных элементов м. б. применены аппараты, дающие спектры длиной 7-15 см. Спектрографы со стеклянной оптикой в общем имеют меньшее значение. Из них удобны комбинированные приборы (например, фирмы Гильгера и Фюсса), которые по желанию можно применять в качестве спектроскопа и спектрографа. Для получения спектров применяются следующие источники энергии. 1) Пламя горящей смеси - водорода и кислорода, смеси кислорода и светильного газа, смеси кислорода и ацетилена или наконец воздуха и ацетилена. В последнем случае температура источника света доходит до 2500-3000°С. Пламя наиболее всего пригодно для получения спектров щелочных и щелочноземельных металлов, а также для таких элементов, как Сu, Hg и Тl. 2) Вольтова дуга . а) Обычная, гл. обр. постоянного тока, силой 5-20 А. С большим успехом она применяется для качественного анализа трудно сплавляемых минералов, которые вводятся в дугу в виде кусочков или тонко растертых порошков. Для количественного анализа металлов применение обычной вольтовой дуги имеет очень существенный недостаток, заключающийся в том, что поверхность анализируемых металлов покрывается пленкой окиси и горение дуги становится в конце концов неравномерным. Температура вольтовой дуги доходит до 5000-6000°С. б) Прерывистая дуга (Abreissbogen) постоянного тока силой 2-5 А при напряжении около 80 V. При помощи специального приспособления горение дуги прерывается 4-10 раз в сек. Этот способ возбуждения уменьшает окисление поверхности анализируемых металлов. При более высоком напряжении - до 220 V и силе тока 1-2 А - прерывистая дуга может применяться также и для анализа растворов. 3) Искровые разряды , получаемые при помощи индукционной катушки или, чаще, трансформатора постоянного или (предпочтительнее) переменного тока мощностью до 1 kW, дающего во вторичной цепи 10000-30000 V. Применяются три типа разрядов, а) Искровые разряды без емкости и индуктивности во вторичной цепи, называемые иногда дугой высокого напряжения (Hochspannungsbogen). Анализ жидкостей и расплавленных солей при помощи таких разрядов отличается большой чувствительностью. б) Искровые разряды с емкостью и индуктивностью во вторичной цепи, часто называемые также конденсированными искрами , представляют собой более универсальный источник энергии, пригодный для возбуждения спектров почти всех элементов (кроме щелочных металлов), а также газов. Схема включения дана на фиг. 1,

где R - реостат в первичной цепи, Тr- трансформатор переменного тока, С 1 - емкость во вторичной цепи I, S - переключатель для изменения индуктивности L 1 , U - синхронный прерыватель, LF - искрогаситель, F - рабочий искровой промежуток. В резонанс ко вторичной цепи I при помощи индуктивности и переменной емкости С 2 настраивается вторичная цепь II; признаком наличия резонанса является наибольшая сила тока, показываемая миллиамперметром А. Назначение вторичной цепи II синхронного прерывателя U и искрогасителя LF - делать электрические разряды возможно однообразными как по характеру, так и по числу в течение определенного промежутка времени; при обычных работах такие добавочные приспособления не вводятся.
При исследованиях металлов во вторичной цепи применяется ёмкость 6000-15000 см и индуктивность до 0,05-0,01 Н. Для анализа жидкостей во вторичную цепь иногда вводится водяной реостат с сопротивлением до 40000 Ом. Газы исследуются без индуктивности с небольшой емкостью. в) Разряды токов Тесла, которые осуществляются при помощи схемы, изображенной на фиг. 2,

где V - вольтметр, А - амперметр, Т - трансформатор, С - емкость, Т-Т - трансформатор Тесла, F - искровой промежуток, куда вводится анализируемое вещество. Токи Тесла применяются для исследований веществ, которые имеют невысокую точку плавления: различных растительных и органических препаратов, осадков на фильтрах и т. п. При спектральном анализе металлов в случае большого их количества они обычно сами являются электродами, причем им придается какая-либо форма, например, из указанных на фиг. 3,

где а - электрод из анализируемой толстой проволоки, b - из жести, с - согнутая тонкая проволока, d - диск, отрезанный от толстого цилиндрического стержня, е - форма, выпиливаемая из больших кусков литья. При количественном анализе необходимо иметь всегда одинаковую форму и размеры подвергающейся действию искр поверхности электродов. При небольшом количестве анализируемого металла можно воспользоваться оправой из какого-либо чистого металла, например, из золота и платины, в которой укрепляется анализируемый металл, как показано на фиг. 4.
Для введения в источник света растворов предложено довольно много способов. При работе с пламенем применяется распылитель Люндегорда, схематически изображенный на фиг. 5 вместе со специальной горелкой.

Продуваемый через распылитель ВС воздух захватывает испытуемую жидкость, наливаемую в количестве 3 -10 см 3 в углубление С, и в виде тонкой пыли относит ее в горелку А, где происходит смешение с газом. Для введения растворов в дугу, а также в искру применяются чистые угольные или графитовые электроды, на одном из которых делается углубление. Необходимо, однако, отметить, что очень трудно приготовить угли совершенно чистыми. Применяемые для очистки способы - попеременное кипячение в соляной и плавиковой кислотах, а также прокаливание в атмосфере водорода до 2500-3000°С - не дают углей, свободных от примесей, остаются (хотя и следы) Са, Mg, V, Ti, Al, Fe, Si, В. Удовлетворительной чистоты получаются также угли путем прокаливания их на воздухе при помощи электрического тока: через угольный стержень диаметром 5 мм пропускается ток силой около 400 А, и достигаемое таким путем сильное накаливание (до 3 000°С) оказывается достаточным для того, чтобы в течение нескольких секунд большинство загрязняющих угли примесей улетучилось. Существуют также такие способы введения растворов в искру, где сам раствор является нижним электродом, и искра проскакивает на его поверхность; другим электродом может служить какой-либо чистый металл. Примером такого устройства может служить изображенный на фиг. 6 жидкостный электрод Герляха.

Углубление, куда наливается испытуемый раствор, облицовывается платиновой фольгой или покрывается толстым слоем позолоты. На фиг. 7 изображен аппарат Хитчена, служащий также для введения растворов в искру.

Из сосуда А испытуемый раствор слабой струей поступает через трубку В и кварцевую насадку С в сферу действия искровых разрядов. Нижний электрод, впаянный в стеклянную трубку, прикрепляется к аппарату при помощи каучуковой трубки Е. Насадка С, изображенная на фиг. 7 отдельно, имеет с одной стороны вырез для стенания раствора. D - стеклянный предохранительный сосуд, в котором делается круглое отверстие для выхода ультрафиолетовых лучей. Сосуд этот удобнее делать кварцевым без отверстия. К верхнему электроду F, графитовому, угольному или металлическому, также приспосабливается предохраняющая от брызг пластинка. Для «дуги высокого напряжения», сильно накаливающей анализируемые вещества, Герлях при работе с растворами применяет электроды с охлаждением, как это схематически показано на фиг. 8.

На толстой проволоке (диаметром 6 мм) укрепляется при помощи пробки К стеклянная воронка G, куда помещаются кусочки льда. На верхнем конце проволоки укрепляется круглый железный электрод Е диаметром 4 см и высотой 4 см, на который накладывается платиновая чашечка Р; последняя должна легко сниматься для очистки. Верхний электрод также д. б. толстым во избежание расплавления. При анализе небольших количеств веществ - осадков на фильтрах, различных порошков и т. д. - можно пользоваться приспособлением, изображенным на фиг. 9.
Из испытуемого вещества и фильтровальной бумаги делается комочек, смачивается для лучшей проводимости раствором, например, NaCl, помещается на нижний электрод, состоящий иногда из чистого кадмия, заключенного в кварцевой (хуже стеклянной) трубочке; верхний электрод также является каким-либо чистым металлом. Для таких же анализов при работе с токами Тесла применяется специальная конструкция искрового промежутка, изображенная на фиг. 10 а и б.

В круглом шарнире К укрепляется в нужном положении алюминиевая пластинка Е, на которую накладывается стеклянная пластинка G, а на последнюю - препарат Р на фильтровальной бумаге F. Препарат смачивается какой-либо кислотой или раствором соли. Вся эта система представляет небольшой конденсатор. Для исследования газов применяются закрытые стеклянные или кварцевые сосуды (фиг. 11).

Для количественного анализа газов удобно пользоваться золотыми или платиновыми электродами, линии которых можно применить для сравнения. Почти все из упомянутых выше приспособлений для введения веществ в искру и дугу при работе укрепляются в специальных штативах. Примером может являться штатив Грамона, изображенный на фиг. 12:

при помощи винта D электроды одновременно раздвигаются и сдвигаются; винт Е служит для передвигания верхнего электрода параллельно оптической скамье, а винт С - для боковых поворотов нижнего электрода; для боковых поворотов всей верхней части штатива служит винт В; наконец при помощи винта А можно поднимать или опускать всю верхнюю часть штатива; Н - подставка для горелок, стаканов и пр. Выбор источника энергии для той или иной цели исследования можно сделать, руководствуясь следующей примерной таблицей.

Качественный анализ . При качественном спектральном анализе открытие какого-либо элемента зависит от многих факторов: от характера определяемого элемента, источника энергии, разрешающей способности спектрального аппарата, а также от чувствительности фотографических пластинок. Относительно чувствительности анализа можно сделать следующие указания. При работе с искровыми разрядами в растворах можно открывать 10 -9 -10 -3 %, а в металлах 10 -2 -10 -4 % исследуемого элемента; при работе с вольтовой дугой пределы открытия лежат около 10 -3 %. Абсолютное количество, которое м. б. открыто при работе с пламенем, составляет 10 -4 -10 -7 г, а при искровых разрядах 10 -6 -10 -8 г исследуемого элемента. Наибольшая чувствительность открытия относится к металлам и металлоидам - В, Р, С; меньше чувствительность для металлоидов As, Se и Те; галоиды, а также S, О, N в их соединениях совсем не м. б. открыты и м. б. открыты лишь в некоторых случаях в газовых смесях.
Для качественного анализа наибольшее значение имеют «последние линии», и при анализе задача заключается в наиболее точном определении длин волн спектральных линий. При визуальных исследованиях длины волн отсчитываются по барабану спектрометра; эти измерения можно считать лишь приблизительными, так как точность составляет обычно ±(2-З) Ӑ и в таблицах Кайзера этому интервалу ошибок могут отвечать около 10 спектральных линий, принадлежащих различным элементам, для λ 6000 и 5000 Ӑ и около 20 спектральных линий для λ ≈ 4000 Ӑ. Гораздо точнее определяется длина волн при спектрографическом анализе. В этом случае на спектрограммах при помощи измерительного микроскопа измеряется расстояние между линиями с известной длиной волны и определяемой; по формуле Гартмана находится длина волны последней. Точность таких измерений при работе с прибором, дающим полоску спектра длиной около 20 см, составляет ± 0,5 Ӑ для λ ≈ 4000 Ӑ, ± 0,2 Ӑ для λ ≈ 3000 Ӑ и ± 0,1 Ӑ для λ ≈ 2500 Ӑ. По длине волны в таблицах находят соответствующий элемент. Расстояние между линиями при обычных работах измеряется с точностью до 0,05-0,01 мм. Этот прием иногда удобно комбинировать со съемками спектров с так называемыми заслонками Гартмана, два типа которых изображены на фиг. 13, а и b; при помощи их щель спектрографа можно делать различной высоты. Фиг. 13, с схематически изображает случай качественного анализа вещества X - установление в нем элементов А и В. Спектры фиг. 13, d показывают, что в веществе Y кроме элемента А, линии которого обозначены буквой G, имеется примесь, линии которой обозначены z. При помощи этого приема в простых случаях можно выполнить качественный анализ, не прибегая к промеру расстояний между линиями.

Количественный анализ . Для количественного спектрального анализа наибольшее значение имеют линии, обладающие возможно большей концентрационной чувствительностью dI/dK, где I - интенсивность линии, а К - концентрация дающего ее элемента. Чем больше концентрационная чувствительность, тем точнее анализ. С течением времени разработан целый ряд методов количественного спектрального анализа. Эти методы следующие.
I. Спектроскопические методы (без фотографической съемки) почти все являются фотометрическими методами. Сюда относятся: 1) Метод Барратта . Одновременно возбуждаются спектры двух веществ - испытуемого и стандартного - видные в поле зрения спектроскопа рядом, один над другим. Ход лучей изображен на фиг. 14,

где F 1 и F 2 - два искровых промежутка, свет от которых проходит через призмы Николя N 1 и N 2 , поляризующие лучи во взаимно перпендикулярных плоскостях. При помощи призмы D лучи попадают в щель S спектроскопа. В его зрительной трубе помещается третья призма Николя - анализатор, - вращая которую добиваются одинаковой интенсивности двух сравниваемых линий. Предварительно при исследованиях стандартов, т. е. веществ с известным содержанием элементов, устанавливается зависимость между углом поворота анализатора и концентрацией, и по этим данным вычерчивается диаграмма. При анализе по углу поворота анализатора из этой диаграммы находится искомое процентное содержание. Точность метода ±10 %. 2) . Принцип метода заключается в том, что лучи света после призмы спектроскопа проходят через призму Волластона, где расходятся на два пучка и поляризуются во взаимно перпендикулярных плоскостях. Схема хода лучей показана на фиг. 15,

где S - щель, Р - призма спектроскопа, W - призма Волластона. В поле зрения получаются два спектра B 1 и В 2 , лежащие рядом, друг над другом; L - лупа, N - анализатор. Если вращать призму Волластона, то спектры будут передвигаться относительно друг друга, что позволяет совместить какие-либо две их линии. Например, если анализируется железо, содержащее ванадий, то совмещается линия ванадия с какой-либо близлежащей одноцветной линией железа ; затем, поворачивая анализатор, добиваются одинаковой яркости этих линий. Угол поворота анализатора, как и в предыдущем методе, является мерой концентрации искомого элемента. Метод особенно пригоден для анализа железа, спектр которого имеет много линий, что позволяет всегда найти линии, пригодные для исследований. Точность метода ± (3-7)%. 3) Метод Оккиалини . Если расположить электроды (например, анализируемые металлы) горизонтально и проектировать изображен из источника света на вертикальную щель спектроскопа, то как при искровых, так и при дуговых разрядах линии примесей м. б. открыты в зависимости от концентрации на большем или меньшем расстоянии от электродов. Источник света проектируется на щель при помощи специальной линзы, снабженной микрометрическим винтом. При анализе эта линза передвигается и вместе с ней передвигается изображение источника света до тех пор, пока какая-либо линия примеси в спектре исчезнет. Мерой концентрации примеси является отсчет по шкале линзы. В настоящее время этот метод разработан также и для работ с ультрафиолетовой частью спектра. Надо отметить, что таким же способом освещения щели спектрального аппарата пользовался Локиер и им был разработан метод количественного спектрального анализа, т. н. метод «длинных и коротких линий». 4) Прямое фотометрирование спектров . Описанные выше методы носят название визуальных. Люндегорд вместо визуальных исследований пользовался для измерения интенсивности спектральных линий фотоэлементом. Точность определения щелочных металлов при работе с пламенем достигала ± 5%. При искровых разрядах этот способ неприменим, так как они менее постоянны, чем пламя. Существуют также способы, основанные на изменении индуктивности во вторичной цепи, а также использующие искусственное ослабление света, попадающего в спектроскоп, до исчезновения в поле зрения исследуемых спектральных линий.
II. Спектрографические методы . При этих методах исследуются фотографические снимки спектров, причем мерой интенсивности спектральных линий является почернение, даваемое ими на фотографической пластинке. Интенсивность оценивается или глазом, или фотометрически.
А . Методы без применения фотометрии . 1) Метод последних линий . При изменении концентрации какого-либо элемента в спектре изменяется число его линий, что дает возможность при неизменных условиях работы судить о концентрации определяемого элемента. Фотографируется ряд спектров веществ с известным содержанием интересующего компонента, на спектрограммах определяется число его линий и составляются таблицы, в которых указывается, какие линии видны при данных концентрациях. Эти таблицы служат дальше для аналитических определений. При анализе на спектрограмме определяется число линий интересующего элемента и по таблицам находится процентное содержание, причем метод дает не однозначную его цифру, а границы концентраций, т. е. «от-до». Наиболее достоверно возможно различить концентрации, отличающиеся друг от друга в 10 раз, например, от 0,001 до 0,01%, от 0,01 до 0,1% и т. д. Аналитические таблицы имеют значение лишь для вполне определенных условий работы, которые в различных лабораториях могут очень сильно различаться; кроме того, требуется тщательное соблюдение постоянства условий работы. 2) Метод сравнительных спектров . фотографируется несколько спектров анализируемого вещества А + х% В, в котором определяется содержание х элемента В, и в промежутках между ними на той же фотографической пластинке -спектры стандартных веществ А + а% В, А + b% В, А + с% В, где а, b, с - известное процентное содержание В. На спектрограммах по интенсивности линий В определяется, между какими концентрациями заключается значение х. Критерием постоянства условий работы является равенство интенсивности на всех спектрограммах какой-либо близлежащей линии А. При анализе растворов в них добавляется одинаковое количество какого-либо элемента, дающего линию близко к линиям В, и тогда о постоянстве условий работы судят по равенству интенсивности этих линий. Чем меньше разница между концентрациями а, Ь, с, … и чем точнее достигнуто равенство интенсивности линий А, тем точнее анализ. А. Райс, например, применял концентрации а, b, с, ... , относящиеся друг к другу, как 1: 1,5. К методу сравнительных спектров примыкает метод «подбора концентраций» (Testverfahren) по Гюттигу и Турнвальду, применимый только к анализу растворов. Он заключается в том, что если в двух растворах, содержащих а% А и х% А (х больше или меньше а), что сейчас же можно определить по их спектрам, то прибавляют в какой-либо из этих растворов такое количество n элемента А, чтобы интенсивность его линий на обоих спектрах стала одинаковой. Тем самым определится концентрация х, которая будет равна (а ± n)%. Можно также прибавить в анализируемый раствор какой-либо другой элемент В до равенства интенсивности определенных линий А и В и по количеству В оценить содержание А. 3) Метод гомологических пар . В спектре вещества А + а% В линии элементов А и В не являются одинаково интенсивными и, если этих линий достаточное количество, можно найти две такие линии А и В, интенсивность которых будет одинакова. Для другого состава А + b% В одинаковыми по интенсивности будут другие линии А и В и т. д. Эти две одинаковые линии называются гомологическими парами. Концентрации В, при которых осуществляется та или иная гомологическая пара, называются фиксирующими пунктами этой пары. Для работы по этому методу требуется предварительное составление таблиц гомологических пар при помощи веществ известного состава. Чем полнее таблицы, т. е. чем больше они содержат гомологических пар с фиксирующими пунктами, отличающимися как можно меньше друг от друга, тем точнее анализ. Этих таблиц составлено довольно большое количество, причем они могут иметь применение в любой лаборатории, т. к. точно известны условия разрядов при их составлении и эти условия м. б. совершенно точно воспроизведены. Достигается это при помощи следующего простого приема. В спектре вещества А + а% В выбираются две линии элемента А, интенсивность которых очень сильно меняется в зависимости от величины самоиндукции во вторичной цепи, именно одна дуговая (принадлежащая нейтральному атому) и одна искровая линия (принадлежащая иону). Эти две линии называются фиксирующей парой . Путем подбора величины самоиндукции линии этой пары делаются одинаковыми и составление ведется именно при этих условиях, всегда указываемых в таблицах. При таких же условиях проводится и анализ, и по осуществлению той или иной гомологической пары находится процентное содержание. Имеется несколько модификаций метода гомологических пар. Из них главнейшим является метод вспомогательного спектра , применяемый в том случае, когда элементы А и В не обладают достаточным количеством линий. В этом случае линии спектра элемента А определенным образом связываются с линиями другого, более пригодного элемента G, и роль А начинает играть элемент G. Метод гомологических пар разработан Герляхом и Швейтцером. Он применим как к сплавам, так и к растворам. Его точность в среднем около ±10%.
В . Методы с применением фотометрии . 1) Метод Барратта . Фиг. 16 дает представление о методе.

F 1 и F 2 - два искровых промежутка, при помощи которых одновременно возбуждаются спектры стандартного и анализируемого вещества. Свет проходит через 2 вращающихся сектора S 1 и S 2 и при помощи призмы D образует спектры, которые расположены один над другим. Путем подбора вырезок секторов линии исследуемого элемента получают одинаковую интенсивность; концентрация определяемого элемента вычисляется из соотношения величин вырезок. 2) является аналогичным, но с одним искровым промежутком (фиг. 17).

Свет от F разделяется на два пучка и проходит через секторы S 1 и S 2 , при помощи ромба Гюфнера R две полоски спектра получаются одна над другой; Sp - щель спектрографа. Вырезки секторов изменяются до получения равенства интенсивности линии примеси и какой-либо близлежащей линии основного вещества и по соотношению величин вырезок высчитывается %-ное содержание определяемого элемента. 3) При применении в качестве фотометра вращающегося логарифмического сектора линии получают на спектрограммах клинообразный вид. Один из таких секторов и его положение относительно спектрографа при работе изображены на фиг. 18, а и б.

Вырезка сектора подчиняется уравнению
- lg Ɵ = 0,3 + 0,2l
где Ɵ - длина дуги в частях полной окружности, находящаяся на расстоянии I, измеренном в мм по радиусу от его конца. Мерой интенсивности линий является их длина, т. к. с изменением концентрации элемента длина его клинообразных линий также изменяется. Предварительно по образцам с известным содержанием строится диаграмма зависимости длины какой-либо линии от %-ного содержания; при анализе на спектрограмме измеряется длина той же линии и по диаграмме находится процентное содержание. Имеется несколько различных модификаций этого метода. Следует указать на модификацию Шейбе, применявшего т. н. двойной логарифмический сектор. Вид этого сектора изображен на фиг. 19.

Линии исследуются затем при помощи специального аппарата. Точность, достижимая при помощи логарифмических секторов, ±(10-15)%; модификация Шейбе дает точность ±(5-7)%. 4) Довольно часто применяется фотометрирование спектральных линий при помощи свето- и термоэлектрических спектрофотометров самых различных конструкций. Удобными являются термоэлектрические фотометры, выработанные специально для целей количественного анализа. Для примера на фиг. 20 приведена схема фотометра по Шейбе:

L– постоянный источник света с конденсором К, М – фотографическая пластинка с исследуемым спектром, Sp - щель, О 1 и О 2 - объективы, V - затвор, Th - термоэлемент, который присоединяется к гальванометру. Мерой интенсивности линий является отклонение стрелки гальванометра. Реже пользуются саморегистрирующими гальванометрами, дающими запись интенсивности линий в виде кривой. Точность анализа при применении этого типа фотометрии составляет ±(5-10)%. При сочетании с другими методами количественного анализа точность м. б. повышена; так, например, метод трех линий Шейбе и Шнеттлера, являющийся сочетанием метода гомологических пар и фотометрических измерений, в благоприятных случаях может дать точность ±(1-2)%.

Мясо по-французски - как много в этом слове для сердца русского слилось… Несмотря на название, это блюдо уже давно стало...
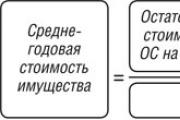
20.09.2017 Вещи, не относящиеся к недвижимости (включая деньги и ценные бумаги), признаются движимым имуществом....

Салат с черносливом относится к тем блюдам, которые подойдут для любого праздника. Их можно приготовить и на Рождество, и...

Для того чтобы разобраться в особенностях рельефа, необходимо знать геологическую историю его формирования. Ученые,...

Креветки вкусны сами по себе без дополнительных ингредиентов, и истинные гурманы советуют наслаждаться ими в качестве...

Куриный рулет с яйцом по Дюкану - замечательное блюдо для того, чтобы скинуть несколько лишних килограммчиков, особенно с...

КАЗАНСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ КАФЕДРА МЕНЕДЖМЕНТА Курсовая работа по теме «Управление временем....
В нашей стране существуют различные виды налогов и имущественный налог - это один из них. Из самого названия...

Цель занятия: изучаем букву Ф, формирование навыка чтения, развитие речевого умения, совершенствование...
Биологическое окисление – это совокупность окислительно-восстановительных реакций, происходящих в живых...
МОЛЕКУЛЯРНЫЙ ВЕС есть относительный вес молекулы вещества. Кроме возможности находиться в трех различных фазах...

Что входит в обязанности диспетчера? Прежде всего - осуществление доставки, формирование производства и многое...

Шницель из говядины на сковороде можно смело назвать универсальным блюдом. Ну, вот посудите сами: шницель...

Готовый продукт получается очень сочным и мягким. Возможно, именно это обстоятельство и является подспорьем для...
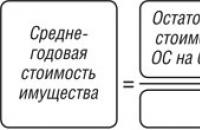
20.09.2017 Вещи, не относящиеся к недвижимости (включая деньги и ценные бумаги), признаются движимым...

Салат с черносливом относится к тем блюдам, которые подойдут для любого праздника. Их можно приготовить и на...